11.5.1. Metadata profile for transcriptomics¶

11.5.1.1. Main Objectives:¶
The main purpose of this recipe is:
To provide guidance on the minimum set of metadata and semantics required to describe any transcriptomics experiments, from standard case-control to dosage-response designs, and from microarrays to single cell RNA sequencing.
11.5.1.1.1. Who is this recipe aimed at?¶
This document is aimed at anyone interested in the metadata, and specifically the ontology annotations, required to capture variables and experimental factors related to a transcriptomics experiment. General knowledge of transcriptomics experiments would be beneficial. No specific technical knowledge is required.
11.5.1.2. Transcriptomics Data model¶
Large sections of any transcriptomics data model are not unique to transcriptomics experiments but are common to a wide range of experiments in the biomedical domain. This includes general project- and sample-level information, which will be briefly covered in this recipe but will also be covered in more depth elsewhere.
Where possible, metadata should be mapped to ontologies to maximise the FAIRness of any dataset. This recipe will suggest possible ontologies for metadata fields where these are available. These lists may however not be exhaustive as new ontologies emerge regularly.
11.5.1.2.1. Existing standards and checklists¶
A set of well-established standards and minimum metadata checklists exist for various aspects of transcriptomics. They include:
Minimum Information About a Microarray Experiment (MIAME) - MIAME is intended to specify all the information necessary for an unambiguous interpretation of a microarray experiment, and potentially to reproduce it. MIAME defines the content but not the format for this information. The MIAME standard has been in existence for over 20 years and has been widely adopted across the scientific community. The data models of the major transcriptomics repositories such as ArrayExpress, the Expression Atlas and the Gene Expression Omnibus (GEO) are MIAME-compliant.
Minimum Information about a high-throughput nucleotide SEQuencing Experiment (MINSEQE) - MINSEQE describes the minimum metadata that is needed to enable the unambiguous interpretation and facilitate reproduction of the results of the experiment. By analogy to the MIAME guidelines for microarray experiments, adherence to the MINSEQE guidelines will improve integration of multiple experiments across different modalities, thereby maximising the value of high-throughput research. MINSEQE has been integrated into a number of transcriptomics and sequencing archives.
Functional Annotation of Animal Genomes (FAANG) - FAANG provides a set of orthogonal standards for the capture of well-structured metadata for experiments, samples and analyses in the animal genomics domain. The FAANG standards support the MIAME and MINSEQE guidelines, and aim to convert them to a concrete specification.
Human Cell Atlas Metadata (HCA-Metadata) - the HCA metadata model provides a concrete specification for capturing the metadata for single cell sequencing experiments. It is based on the three standards above, while focusing on the specific requirements of the single cell space.
11.5.1.2.2. Minimum metadata vs desirable metadata¶
A minimum metadata set constitutes metadata items that always need to be supplied with any experimental data. For validation purposes, these should not be omittable. Desirable or recommended metadata items on the other hand should be supplied if available but will not cause a validation failure if absent.
11.5.1.2.3. Common metadata¶
Common metadata include any information that is not specific to transcriptomics experiments but applies to most experiments in the biomedical domain. They include:
Project level metadata
Common sample level metadata such as species, tissue, cell type etc.
11.5.1.2.3.1. Suggested metadata fields¶
The following table contains a non-exhaustive list of suggested minimum metadata fields for biological samples. The collection is based on a range of existing metadata standards, including MIAME, MINSEQE, FAANG and HCA. Fields were included if they occurred in at least two of the standards.
Metadata field |
Required? |
Definition |
Comment |
|---|---|---|---|
unique ID |
required |
Identifier for a sample that is at least unique within the project |
|
sample type |
required |
The type of the collected specimen, eg tissue biopsy, blood draw or throat swab |
|
species |
required |
The primary species of the specimen, preferably the taxonomic identifier |
This may not be the same as the “host” organism, eg in the case of a PDX tissue sample, the host may be a mouse but the tissue may be human. Ontology field - NCBITaxonomy |
tissue/organism part |
required |
The tissue from which the sample was taken |
|
disease |
required |
Any diseases that may affect the sample |
This may not necessarily be the same as the host’s disease, eg healthy brain tissue might be collected from a host with type II diabetes while cirrhotic liver tissue might be collected from an otherwise healthy individual. Ontology field - e.g. MONDO or DO |
sex |
required |
The biological/genetic sex of the sample |
|
development stage |
required |
The developmental stage of the sample |
|
collection date |
required |
The date on which the sample was collected, in a standardised format |
Collection date in combination with other fields such as location and disease may be sufficient to de-anonymise a sample |
external accessions |
recommended |
Accession numbers from any external resources to which the sample was submitted |
eg Biosamples, Biostudies |
strain |
recommended |
Strain of the species from which the sample was collected, if applicable |
|
ancestry/ethnicity |
recommended |
Ancestry or ethnic group of the individual from which the sample was collected |
|
age |
recommended |
Age of the organism from which the sample was collected |
|
age unit |
recommended |
Unit of the value of the age field |
|
BMI |
recommended |
Body mass index of the individual from which the sample was collected |
Only applies to human samples |
treatment category |
recommended |
Treatments that the sample might have undergone after collection |
|
cell type |
recommended |
The cell type(s) known or selected to be present in the sample |
|
growth conditions |
recommended |
Features relating to the growth and/or maintenance of the sample |
|
genetic variation |
recommended |
Any relevant genetic differences from the specimen or sample to the expected genomic information for this species, eg abnormal chromosome counts, major translocations or indels |
|
sample collection technique |
recommended |
The technique used to collect the specimen, eg blood draw or surgical resection |
|
phenotype |
recommended |
Any relevant (usually abnormal) phenotypes of the specimen or sample |
|
cell cycle |
recommended |
The cell cycle phase of the sample (for synchronized growing cells or a single-cell sample), if known |
|
cell location |
recommended |
The cell location from which genetic material was collected (usually either nucleus or mitochondria) |
|
11.5.1.2.4. Assay metadata¶
Assay-level metadata covers any metadata directly related to the preparation of the biomaterial undergoing the assay and the process of performing the assay. Most, though not all, of this metadata is specific to transcriptomics. Examples include:
Library information, including what sample the library was originally derived from and whether any replicates are biological or technical
Process (wet experiment) metadata
Technology type (e.g. bulk RNA-Seq, scRNA-Seq, CITE-Seq)
Instrument metadata
Other assay-specific metadata
QC information
Workflow metadata
The figure below shows an excerpt of the Human Cell Atlas json schema for describing the process of sequencing.

Fig. 11.2 human cell atlas sequencing json schema.¶
Note
Note the key attributes for which controlled vocabulary may be directly specified as a:
json schema ENUM
as found at line 79, or may be specified via a reference to a dedicated ontology schema as in:
"$ref": "module/ontology/biological_macromolecule_ontology.json"
as found at line 82.
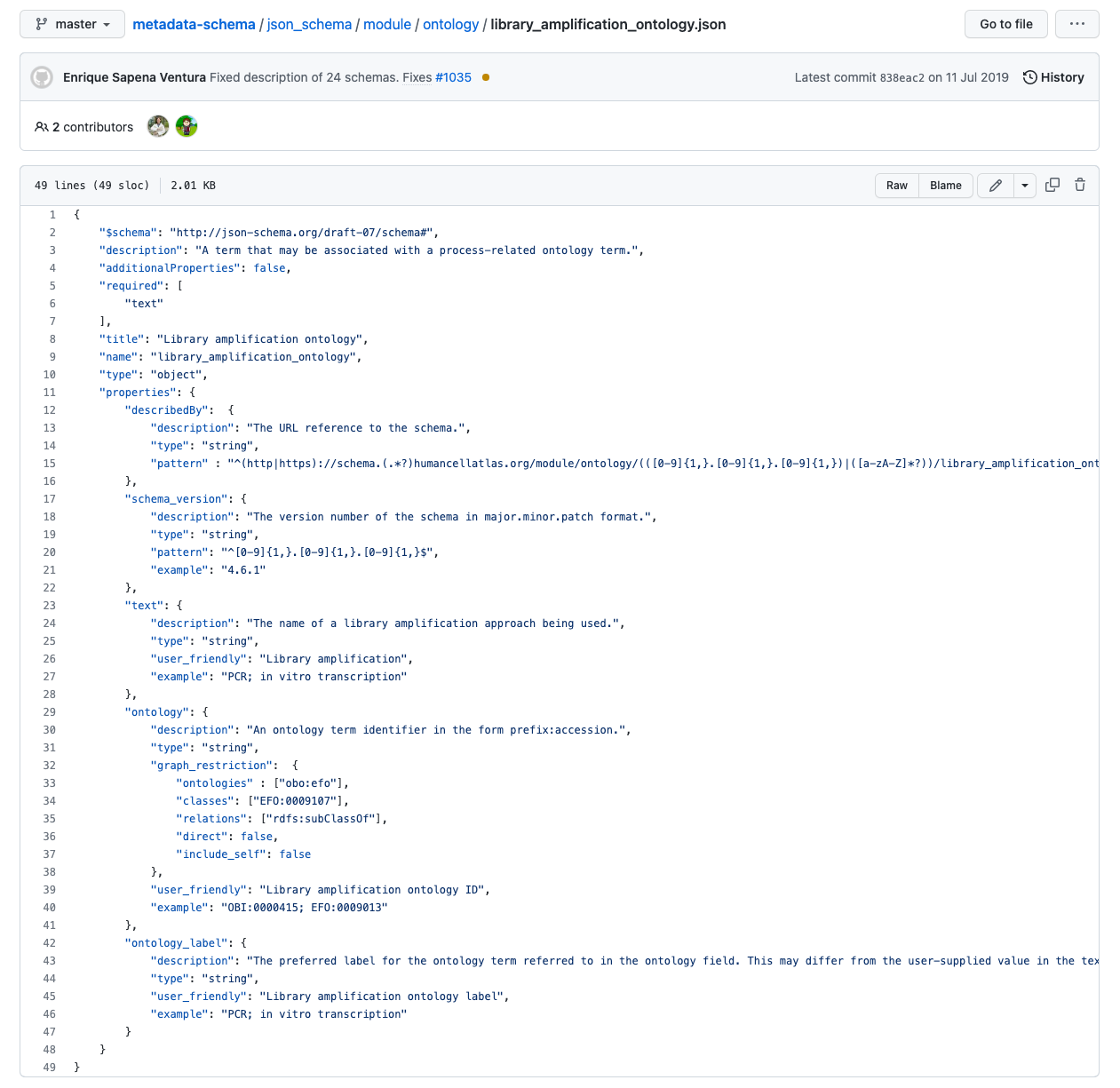
Fig. 11.3 human cell atlas sequencing json schema.¶
Note
Note how the ontology element defines a graph restriction pointing to a branch in a semantic resource, (EFO in this case).
11.5.1.2.4.1. Suggested metadata fields¶
The following table contains a non-exhaustive list of suggested minimum metadata fields for assays. The collection is based on a range of existing metadata standards, including MIAME, MINSEQE and HCA. This list can and should be further broken down based on specific technologies used, such as microarrays or whole genome sequencing.
Metadata field |
Required? |
Definition |
Comment |
|---|---|---|---|
unique ID |
required |
Identifier for the assay that is at least unique within the project |
|
experiment type |
required |
The type of experiment performed, eg ATAC-seq or seqFISH |
|
extracted nucleic acid/material type |
required |
The type of material that was extracted from the sample, eg polyA RNA |
|
platform |
required |
The type of instrument used to perform the assay, eg Illumina HiSeq 4000 or Fluidigm C1 microfluidics platform |
|
nucleic acid extraction method |
required |
Technique used to extract the nucleic acid from the cell |
|
cDNA library amplication method |
required |
Technique used to amplify a cDNA library |
|
array or sequencing method |
required |
The array or sequencing technology used - may be the same as |
|
biological or technical replicate |
required |
Information whether the sample on which the assay was performed was biological or technical replicate. |
boolean or CV |
end bias |
required |
The type of tag or end bias the library has, eg 3 prime tag or 5 prime end bias |
standardised field or ontology |
external accessions |
recommended |
Accession numbers from external resources to which assay or protocol information was submitted |
eg protocols.io, AE |
instrument model |
required |
The specific instrument on which the assay was performed. Essential for QC purposes. |
|
assay start time |
recommended |
The exact time at which the assay was started |
|
assay end time |
recommended |
The exact time at which the assay was completed |
|
assay duration |
recommended |
The duration, in a relevant time unit (eg minutes or hours), of the assay from start to finish |
|
array quality |
recommended |
The overall quality of the array |
|
chemical compound |
recommended |
Any relevant chemical compounds used in the assay |
|
labeling molecule used |
recommended |
The type of labeling molecule used in an array-based experiment |
|
spike-in kit used |
recommended |
Information about the spike-in kit used during sequencing library preparation |
|
cDNA primer |
recommended |
Type of primer used for cDNA synthesis from RNA, eg polyA or random |
standardised field or ontology |
library strandedness |
recommended |
The strandedness of the cDNA library |
standardised field or ontology |
cell quality |
recommended |
Information about the quality of a single cell such as morphology or percent viability |
standardised field or ontology |
cell barcode |
recommended |
Information about the cell identifier barcode used to tag individual cells in single cell sequencing |
|
UMI barcode |
recommended |
Information about the Unique Molecular Identifier barcodes used to tag DNA fragments |
11.5.1.2.5. Analysis metadata¶
Analysis-level metadata includes any metadata related to the files that come out of the experiment, from the sequencing or imaging files generated directly by the machine to files generated during the various stages of processing and analysis, as well as details of any analyses performed. It is very important to always capture versions of software and reference genomes used in order to allow accurate replication of results. Analysis metadata includes
Type of analysis
File formats, e.g. BAM, fastq or gene count
File location e.g. URL
Summarisation of data, e.g. enrichment analysis
11.5.1.2.5.1. Suggested metadata fields¶
The following table contains a non-exhaustive list of suggested minimum metadata fields for analyses. The collection is based on a range of existing metadata standards, including MIAME, MINSEQE and HCA. This list can and should be further broken down based on the specific analysis type (primary, secondary or teriatry analysis, meta-analysis etc)
Metadata field |
Required? |
Definition |
Comment |
|---|---|---|---|
analysis type |
required |
The type of analysis performed, eg genome assembly or variant calling |
|
computational method |
required |
The specific computational method or algorithm used as part of the analysis |
|
normalisation strategy |
required |
The approach used to normalise the data |
|
file format |
required |
The file format in which the analysis is provided |
|
file storage location |
required |
The location in which the data files are stored |
|
software package |
recommended |
The software package used for data analysis |
|
software version |
recommended |
The exact version number of the software package |
|
analysis date |
recommended |
The date on which the analysis was performed |
|
read index |
recommended |
The sequencing read a specific file represents, eg read1 or index1 |
|
read length |
recommended |
The length of a sequenced read in this file, in nucleotides. |
|
assembly type |
recommended |
The assembly type of the genome reference file, eg primary, complete or patch assembly. |
standardised field or ontology |
reference genome version |
recommended |
The genome version of the reference file. |
11.5.1.3. Ontologies for transcriptomics data¶
While it is essential that any transcriptomics metadata be annotated with ontology terms wherever possible, there is no absolute set of ontologies that must be used above all others. There is however a consensus set of ontologies and other standardised resources that are commonly used in transcriptomics metadata, including in the main data archives. The most commonly used ontologies and fields they apply to are listed below. This table represents an absolute minimum of ontology annotations that should be included in a transcriptomics metadata set for it to be considered as FAIR. Not all fields suggested for ontology annotation in the previous section are repeated here for this reason.
Data type |
Ontology/Entity sources |
Type |
Notes |
|---|---|---|---|
Species |
NCBI taxonomy Scientific name + ID |
Ontology |
|
Tissue |
Uberon term and Id |
Ontology |
|
Cell type |
CL term and Id |
Ontology |
|
Disease |
MONDO, DO or MeSH |
Ontology |
no single solution - options vary depending on resource and individual requirements |
Phenotype/Trait |
HPO (human), MP(other mammals), various others for model organisms (yeast, zebrafish, Xenopus, C. elegans) |
Ontology |
|
Experiment Type |
EFO, OBI |
Ontology |
e.g. RNASeq, CITESeq etc. - |
Cell location/cycle |
GO |
Ontology |
|
Developmental stage |
HSAPDV/Uberon |
Ontology |
|
Chemical compound |
ChEBI |
Ontology |
|
Chemical compound |
UniChem, SMILE, InChi |
Entity |
|
Gene/protein |
ENSEMBL, ENTREZ_GENE, UNIPROT, HGNC ID, INSDC |
Entity |
|
Metabolites |
MetaboLights compound accession, ChEBI |
Entity |
|
Nucleotide reference sequence |
ReqSeq |
Entity |
11.5.1.4. Conclusion¶
Using common transcriptomics metadata standards, in particular the fields listed above as guidance, it is possible to easily define a comprehensive metadata template to capture all the experimental variables to describe any transcriptomics experiment in a FAIR-compliant way. A generic step-by-step guide for designing a metadata template is provided here
References
11.5.1.4.1. What to read next?¶
FAIRsharing records appearing in this recipe:
- ArrayExpress
- Binary Alignment Map Format (BAM)
- BioStudies
- Cell Ontology (CL)
- Chemical Entities of Biological Interest (ChEBI)
- EMBRACE Data and Methods Ontology (EDAM)
- Ensembl
- Experimental Factor Ontology (EFO)
- Expression Atlas
- Gene Expression Omnibus (GEO)
- Gene Ontology (GO)
- HUGO Gene Nomenclature Committee (HGNC)
- Human Ancestry Ontology (HANCESTRO)
- Human Developmental Stages Ontology (HSAPDV)
- Mammalian Phenotype Ontology (MP)
- MetaboLights (MTBLS)
- Minimal Information about a high throughput SEQuencing Experiment (MINSEQE)
- Minimum Information About a Microarray Experiment (MIAME)
- Monarch Disease Ontology (MONDO)
- NCBI Taxonomy (NCBITAXON)
- NCI Thesaurus (NCIt)
- Ontology for Biomedical Investigations (OBI)
- Ontology for General Medical Science (OGMS)
- The FAIR Principles (FAIR)
- Units Ontology (UO)
- protocols.io
11.5.1.5. Authors¶
Authors
Name |
ORCID |
Affiliation |
Type |
ELIXIR Node |
Contribution |
|---|---|---|---|---|---|
University of Luxembourg |
|
Writing - Review & Editing |
|||
Karsten Quast |
Boehringer-Ingelheim AG |
||||
GSK |